Genetikai betegségek
meghatározás
A genetikai betegség vagy az örökletes betegség olyan betegség, amelyet az érintett egy vagy több génje okoz. A DNS itt a betegség közvetlen kiváltójaként szolgál. A legtöbb genetikai betegség esetében a kórokozó gének elhelyezkedése ismert. Ha genetikai betegség gyanúja merül fel, a megfelelő diagnózist genetikai vizsgálat útján lehet meghatározni.
Másrészről számos olyan betegség is létezik, amelyek előfordulása genetikai hatással bír vagy tárgyalásra kerül, például cukorbetegség („cukorbetegség”), csontritkulás vagy depresszió. Ez úgynevezett dispozíció, azaz bizonyos betegségek megnövekedett valószínűsége. A diszpozíciókat meg kell különböztetni az örökletes betegségektől.

Ezek gyakori örökletes betegségek
Abszolút értelemben az örökletes betegségek nem gyakoriak, de az itt felsorolt örökletes betegségek gyakran előfordulnak, összehasonlítva a genetikai okú egyéb betegségekkel.
-
Marfan-szindróma
-
Sarlósejtes vérszegénység
-
Hemophilia (A vagy B hemofília)
-
V faktor Leiden mutáció és az abból eredő APC-rezisztencia
-
Piros zöld gyengeség
-
Glükóz-6-foszfát dehidrogenáz hiány (G6PD hiány)
-
Polydactyly ("több ujj", tünetként is lehetséges más betegségek esetén)
-
21. triszomia (Down-szindróma)
-
Chorea Huntington
okoz
Az örökletes betegségek megjelenése rendkívül változatos. Alapvetően csak egy közös dolog van: mindegyikük oka a DNS-ben, azaz az érintett személy genetikai anyagában rejlik. Különféle változások léphetnek fel itt, például mutációk (DNS-információcsere) vagy deléciók (bizonyos genetikai anyag hiánya).
Nagyon sok információ kódolódik a genetikai anyagban, például a test sejtek működése szempontjából fontos különféle komponensek „tervrajzai”. Ezek lehetnek enzimek, elektrolitcsatornák vagy hírvivő anyagok. Ezeket a legkisebb elemeket ezután hibásan olvassa le vagy egyáltalán nem olvassa le a DNS-ből, amely hiányzik a test kifinomult rendszerében. A helytelen vagy hiányzó genetikai információ tehát bizonyos rendellenességeket okoz a testben. Ezek ezután tüneteket okoznak annak a funkcionális rendszernek megfelelően, amelyben egy elem hiányzik.
Itt található a témával kapcsolatos összes információ: A genetikai teszt.
Így öröklik az örökletes betegségeket
Minden örökletes betegséget akár monogenetikusan, akár poligenetesen örökölnek: Ez azt jelenti, hogy egy vagy több genetikai helyet meg kell változtatni a betegség kialakulásához.
Ezenkívül a genetikai tulajdonságok mindig öröklődhetnek domináns vagy recesszív módon: A recesszív azt jelenti, hogy hajlandóságnak kell lennie erre az örökletes betegségre mind az apai, mind az anyai génekben. Domináns öröklés esetén egy változás (azaz egy szülő) elegendő a betegség kiváltásához. Ebből következik, hogy a túlnyomórészt öröklött betegségek esetén a hordozók is megbetegednek - míg a recesszív öröklés esetén általában még azt sem tudják, hogy van-e megfelelő genetikai hajlam.
Vannak olyan betegségek is, amelyek a nemi kromoszómákon keresztül öröklődnek, például hemofília vagy vörös-zöld vakság. Ennek lehetőségei általában az X kromoszómán találhatók, mivel az Y kromoszóma összességében nagyon kicsi, és általában kevés genetikai információt képes tárolni. Ezért az X-hez kapcsolódó örökletes betegségekről beszélünk. Ezek általában sokkal inkább a férfiakat érintik, mint a nőket, mivel a nők a másodikval kompenzálhatják az X kromoszómán szereplő téves információkat.
A genetikai betegség öröklődése általában könnyen felkutatható, ha érdekli.
Vizsgálatok születés előtt
A gyermek genetikai anyaga elvileg már megvizsgálható a méhben minden örökletes betegség szempontjából, amelyek ok-okozati genetikai helyzete ismert. A genetikai elemzések azonban időigényesek, tehát általában csak a feltételezett génhelyet elemezzük - ehhez viszont indokoltnak kell lennie a genetikai betegség gyanújának.
Egy ilyen vizsgálathoz genetikai anyagot vehetünk az amniotikus folyadékból vagy a méhlepéből, és felhasználhatjuk az elemzéshez.
Mindig azonban szem előtt kell tartani, hogy az invazív diagnózis a születendő gyermek életét is veszélyezteti. Az ilyen lyukasztásokat ezért minden esetben külön-külön meg kell mérni.
Vannak olyan mérések is, amelyek genetikai betegségre utalhatnak, például a nhalális átlátszóság mérése a trisomia 21 jeleként. Ezek a módszerek nem veszélyesek a születendő gyermekre, de nem nyújtanak abszolút bizonyosságot a genetikai betegség jelenlétében. Tehát itt is egy műveletet gondosan mérlegelni kell.
21. triszómia
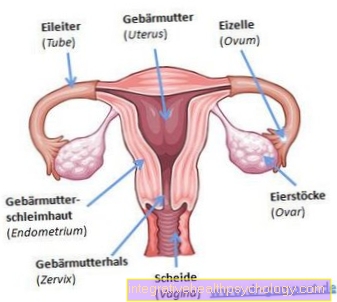
A 21. trisizomia oka a 21. kromoszóma, amely az érintett személyekben nem kétszer, hanem háromszor jelenik meg. A DNS ezen változatát akkor hozzák létre, amikor a kromoszómák eloszlanak a szülői csírasejtekben, azaz a sperma- vagy petesejtekben. Ezért "elosztási hiba", és nem a tényleges genetikai anyag megváltozása. Ez megmagyarázza, hogy miért jelentkezhet spontán módon a 21. sz. Triszómia minden családban, és miért minden családban azonos a Down-szindrómás gyermek esélye. Szigorúan véve, a 21. trizómiát - mint a többi triszómiát - valódi értelemben nem kell örökletes betegségnek tekinteni. Ennek ellenére a 21. trisizomia a leggyakoribb DNS-sel kapcsolatos betegség újszülöttekben.
A Down-szindrómában bekövetkezett megváltozott kromoszómák jellemzői már a méhben lévő születendő gyermekeknél megfigyelhetők: A növekedés késése és rendellenességei többek között túl kicsi koponyához vezethetnek, a comb és a felkar rövid csontjai, valamint a szívhibák. A nagy mennyiségű amniotikus folyadék a 21. trizomia jele is lehet, mivel az érintett születendő gyermekek viszonylag kevés amniotikus folyadékot fogyasztanak vagy nyelnek le. Ezeknek a tulajdonságoknak azonban nincs a Down-szindróma határozott jele!
A fent említett növekedési lassulás jelein kívül a Down-szindrómás gyermekek gyakran mutatnak késleltetett fejlõdést is, például a nyelv és a motoros készségek területén. A Down-szindróma által érintett emberek gyakran figyelemre méltó társadalmi készségeket mutatnak, míg az intelligencia gyakran az átlag alatt marad. Ugyanakkor az érintett emberek nagyban különböznek egymástól ezekben a jellemzőkben, és nem ritka, hogy jó támogatás után az iskolába járnak.
Később az életben a 21-es triszómiában szenvedő embereknek megnövekedett annak kockázata, hogy bizonyos betegségeket diagnosztizálnak. Ide tartoznak az Alzheimer-kór, az epilepszia és a rák, különösen a leukémia. Ennek ellenére a Down-szindrómás emberek várható élettartama tovább növekszik: Időközben az érintett emberek gyakran elérik a 60 vagy 70 évet.
További információt a weboldalunkon talál Down szindróma
Alfa-1 antitripszin hiány
Az alfa-1 antitripszin-hiány különböző formákat ölthet, az érintett személy pontos genetikai tulajdonságaitól függően. Ez azt jelenti, hogy nem minden alfa-1 antitripszin-hiány vezet tünetekhez. Az alábbiakban csak e genetikailag meghatározott betegség klinikailag szembetűnő típusát (PiZZ) tárgyaljuk.
Az ebben a betegségben előforduló enzimhiány az érintett személyekben a szervszövetben építőelemek lebontását és átalakulását idézi elő. Ezenkívül a hibás fehérjéket a máj kiszűri a vérből és ott felhalmozódik. Ez májgyulladáshoz (hepatitisz), cirrhosishoz vagy májrákhoz vezethet. A tüdő légutak instabilvá válnak a stabil szövetek hiánya miatt, és gyorsabban összeomlanak: A COPD (krónikus obstruktív tüdőbetegség) klinikai képe alakul ki. Ez a klinikai kép gyakran az alfa-1 antitripszin-hiány első tünete, ezért minden COPD-vel rendelkező fiatalabb személyt ellenőrizni kell az alfa-1 antitripszin-hiány szempontjából.
Ha a betegség hosszú ideig fennáll, a tüdő túlfújhat, mivel a belélegzett levegőt nem lehet megfelelően kilégzni az instabil légutakon, és felhalmozódik a tüdőben. Terápiaként a cigarettázás és a légzőszervi betegségek megelőzésére szolgáló rendszeres oltások következetes elkerülése mellett gyógyászati intézkedéseket kell hozni: A hiányzó alfa-1-antitripszint intravénásán beadhatják a tünetek lehető legnagyobb mértékű enyhítésére és a betegség lefolyásának megállítására.
További információt a weboldalunkon talál Alfa-1 antitripszin hiány
vérzékenység
A hemofília csoportját nyelvtudással „hemofília” néven ismerik, mivel ez a kifejezés nagyon pontosan leírja ennek az örökletes betegségnek a fő tünetét: az érintett emberek hosszabb ideig vérznek, és a betegség súlyosságától függően gyakrabban, mint érintetlenek.
A vérzést általában az ún. Koagulációs kaszkád, az endogén jelátviteli út állítja le, amely megakadályozza a túlzott vérvesztést. Ebben a koagulációs rendszerben 13 tényező játszik szerepet, amelyek egymás után aktiválódnak. Ezt dominó sorozatként lehet elképzelni: ha megüt egy kőre (véralvadási tényező), akkor aktiválja a következőt és így tovább. Ezen jelút vagy a dominó végén vér koaguláció van. Hemofília esetén egy bizonyos tényező hiányzik - a betegség konkrét altípusától függően: a láncreakció itt szakad meg.
A betegség terápiáját úgy lehet elvégezni, hogy meghatározzák a hiányzó tényezőt és hozzáadják azt kívülről. Az érintett embereket ezért rendszeresen be kell injektálni magukkal a véralvadási tényezővel, hogy a láncreakció fennmaradjon.
További információt a weboldalunkon talál Vérbetegség
Cisztás fibrózis
A genetikai betegségben a cisztás fibrózis - más néven cisztás fibrózis - az ioncsatornák, pontosabban a klorid csatornák hibás előállítása fordul elő. Ennek eredményeként az érintett személyek testváladékainak összetétele (például izzadság, légzőrendszer és hasnyálmirigy kiválasztása) megváltozik: Mivel a klorid hiánya azt jelenti, hogy kevesebb víz kerül a megfelelő mirigy vezetékébe, a szekréció viszonylag viszkózus.
Ennek eredményeként a tünetek általában az emésztőrendszerben alakulnak ki, mivel az emésztő enzimekkel történő szekréció nem képes jól folyni a hasnyálmirigyből a bélbe, és így maga a hasnyálmirigy károsodik. Ezen túlmenően gyakori az emésztőrendszeri rendellenességek, például zsíros széklet, hasmenés és az ebből következő alacsony testtömeg.
A tünetek második nagy csoportja általában a tüdőben alakul ki: Mivel a tüdőben természetesen előforduló nyálkahártya viszkózusabb, mint egészséges embereknél, nehezebb eltávolítani azt a ciliáról. Ez krónikus köhögéshez és a hörgők elzáródásához (bronchectasis) vezethet. A nagyobb tüdőszekréció jó környezetet biztosít a baktériumok növekedéséhez, ami gyakori légúti fertőzéseket és tüdőgyulladást eredményez.
A cisztás fibrózist tünetileg kezelik köpködőkkel, emésztő enzimekkel és antibiotikumokkal fertőzések esetén.
Erről további információt a weboldalunkon találhat Cisztás fibrózis
V faktor Leiden és APC ellenállás
A V faktor Leiden mutációja magában foglalja a genetikai információk megváltozását, amely fokozott vérrögképződést okozhat. Ennek oka a test úgynevezett véralvadási kaszkádjában szereplő V faktor: ez a jelút biztosítja, hogy sérülés esetén a seb a test saját "tapadó fehérjéivel" (fibrin) záródik be. Ebben a jelzőútban 13 tényező található, amelyeket római számokkal nevezünk (ez azt jelenti, hogy „5. tényező szenvedés”!). A V faktor jótékony hatással van a fibrindugó kialakulására, de gátolható az úgynevezett aktivált C protein (röviden APC). Ez fontos szerepet játszik e jelátviteli út szabályozásában és a túlzott vérrögképződés megelőzésében.
A mutáns V faktor jelen van az érintett egyénekben, de nem reagál az APC-re. Ezen a ponton a testnek hiányzik egy fontos „biztonsági eszköz”, amely ok nélkül megakadályozza a véralvadást, amely akár az erek elzárását is okozhatja, és ezzel keringési rendellenességeket okozhat.
Statisztikai szempontból azok az emberek, akiket a V faktor Leiden mutáció érint, valószínűbb, hogy trombózisos eseményeket (azaz trombózist vagy tüdőembólia) szenvednek, még akkor is, ha tipikus kockázati tényezők nem fordultak elő. Technikai szempontból a „trombofíliáról”, azaz a véralvadási hajlamról is beszélünk.
Erről további információt a weboldalunkon találhat V. faktor Leiden
Gaucher-kór
Gaucher-kór esetén a DNS-információ megváltozása a lipid-metabolizmusban részt vevő enzim, pontosabban a glükocerebrosidáz hibáját okozza: Ez elősegíti a régi sejtkomponensek lebontását. Hiba esetén csökkent a funkcionalitás vagy akár a funkcionalitás is veszíthet, ennek megfelelően a tünetek gyermekkorban vagy fiatal felnőttkorban jelentkezhetnek.
A Gaucher-kór tünetei nagyrészt a máj és a lép megnövekedett oka, amelynek növekedése a test megpróbálja kompenzálni az enzimek hiányát. Ez növeli az összes vérkomponens lebontását, amely felismerhető a vérképben és diagnosztikai indikátorként használható a megnövekedett májral és léppel együtt.
A hiányzó glükocerebrosidáz enzim gyógyszerként alkalmazható. A Gaucher-kór előrejelzése és lefolyása nagymértékben függ az enzim funkcióvesztésének súlyosságától.
További információt itt olvashat: Gaucher-kór.
Osler-kór
Az Osler-kór egy örökletes betegség, amelyet erős ér-tágulás jellemez. Az érrendszer ilyen kiterjedése elvileg bárhol megtörténhet, mind a bőrön, mind a belső szerveken. A kibővített edények falai viszonylag vékonyak és könnyen letépnek. Ennek eredményeként az érintett területek gyorsan vérzik.
Az értágítás különösen gyakran fordul elő az arcon és az orr nyálkahártyáján, így az érintett emberek általában gyakori orrvérzésről és az arc kis foltos vérzéséről panaszkodnak.
Osler-kór gyanúja esetén megfelelő diagnosztikát kell elvégezni, mivel az értágítás elõfordulhat olyan létfontosságú szervekben vagy jó vérellátással járó szervekben is, mint például a tüdõ, az agy vagy a máj, amelyekben a felszakadt edényből származó vérzés veszélyes.
A témáról többet találhat weboldalunkon Osler-kór
Recklinghausen-kór
Az 1. típusú neurofibromatózis - vagy a Recklinghausen-kór - olyan genetikai betegség, amelyben az érintett személyeknél daganatok alakulnak ki az idegfedő sejtjein. A kialakuló daganatok lehetnek jóindulatúak és rosszindulatúak, és fiatal korban jelentkezhetnek.
A tipikus daganatok azonban jóindulatú neurofibrómák: Ezek olyan sejtekből állnak, amelyek elektromos kábellel burkolják és izolálják az ideget, valamint a környező kötőszövetből. Ezek jóindulatúak, azaz nem terjednek és lassan növekednek.
A neurofibrómák eltávolítását célzó műtét azonban nehéz lehet, mivel gyakran szorosan kapcsolódnak az ideghez, majd a megfelelő ideget el kell távolítani. Mindazonáltal ez a tüneti neurofibroma egyetlen kezelési lehetősége, mivel ennek az örökletes betegségnek az okozati kezelése nem lehetséges.
A témáról többet találhat weboldalunkon 1. típusú neurofibromatózis
Izomsorvadás
Az izomdisztrófia kifejezés olyan örökletes betegségek egy csoportját írja le, amelyben bizonyos izomkomponenseket a testsejtek nem képesek vagy nem tudnak megfelelően összeállítani. Ennek eredményeként az érintett emberek izomgyengesége általában már gyermekkorban és serdülőkorban alakul ki, és ez izomtömeg elvesztését, mozgáskorlátozásokat és akár testi fogyatékosságot eredményezhet.
Az izomdisztrófia gyanúja esetén először meg kell határozni a vérértéket. Ha az értékek megegyeznek a feltételezett diagnózissal, izom-biopsziát lehet elvégezni: Egy kis szövetmintát veszünk az izomból, amelyet mikroszkóposan megvizsgálunk sejthibák szempontjából. A diagnózis megállapításához genetikai vizsgálat is lehetséges, mivel a megfelelő genetikai helyek általában ismertek az izomdisztrófia különböző formáira, és ezeket meg kell változtatni. Az izomdisztrófiák okozati kezelése nem ismert.
A témáról többet találhat weboldalunkon Izomsorvadás
Xeroderma pigmentosum
A Xeroderma pigmentosum egy ritka örökletes betegség, amelyben az érintett személy bőrén lévő bizonyos enzimek nem működnek. Ezek az enzimek általában a DNS javulását gondozzák, amelyet napfény vagy a benne lévő UVB fény károsíthat. Az UVB károsodás bőrrákot okozhat az érintett emberekben és minden más emberben, de a Xeroderma Pigmentosum esetében a folyamatot javító mechanizmusok hiánya gyorsítja. Ennek eredményeként az érintett emberek gyermekkori és serdülőkori, valamint rövid napsugárzásnak kitett bőrrák súlyos formáit alakítják ki.
Okozati kezelés még nem lehetséges. Az érintett embereknek az élet során el kell kerülniük a napfényt, ezért a „holdfény gyermekek” becenév betelepült az érintett (néha nagyon fiatal) emberek számára. Ezen túlmenően ezeket az embereket dermatológusnak kell felügyelnie a rendszeres bőrrák szűrés céljából, az újonnan kialakult bőrrák azonnali eltávolítása érdekében. Ha ezeket az intézkedéseket szigorúan betartják, akkor a xeroderma pigmentosummal rendelkező személy várható élettartama nagyjából megegyezik egy érintetlen személy élettartamával.
A betegségről további információkat a weboldalunkon találhat Xeroderma pigmentosum
Lynch-szindróma
A Lynch szindróma a DNS olyan változása, amely hibás enzimet okoz a test sejtjeiben.Az érintett emberekben tehát egy bizonyos mechanizmus hibás, amely egyébként feltételezi, hogy megvédi a sejteket a degenerációtól, azaz az ellenőrizetlen növekedéstől - ezért a Lynch-szindrómás embereknél a rák kialakulásának kockázata jelentősen megnövekedett.
A vastagbélrák itt gyakran előfordul, mivel a sejtek természetesen itt is osztódnak, és a sejtek növekedésének és elhalálozásának hibái gyorsabban észlelhetők. Az érintett embereknek a vastagbélben daganat alakul ki szokatlanul fiatal korban, azaz 50 éves kor előtt, ezt HNPCC-nek (örökletes nem polipózis vastagbélrák) hívják. Azonban nem mindenkinek, aki rendelkezik a Lynch-szindróma genetikai felépítésével, alakul ki vastagbélrák. Másrészről más szervek is kialakíthatnak daganatot, mivel a daganat kialakulását elősegítő genetikai hajlandóság minden testsejtben megtalálható. Ezért rendszeres ellenőrzésre és megelőző vizsgálatokra van szükség a Lynch-szindróma által érintett személyek számára a korai stádiumban kialakuló daganatok megfelelő kezelése érdekében.
A témáról többet találhat weboldalunkon Lynch-szindróma